



















Sleep apnea device recall drags on, stoking frustration
Philips devices are coming off of shelves after more than 70,000 reports of problems

WASHINGTON – A massive recall of millions of sleep apnea machines has stoked anger and frustration among patients, and U.S. officials are weighing unprecedented legal action to speed a replacement effort that is set to drag into next year.
Sound-dampening foam in the pressurized breathing machines can break down over time, leading users to potentially inhale tiny black particles or hazardous chemicals while they sleep, manufacturer Philips warned in June 2021.
Philips initially estimated it could repair or replace the units within a year. But with the recall expanding to more than 5 million devices worldwide, the Dutch company now says the effort will stretch into 2023.
That’s left many patients to choose between using a potentially harmful device or trying risky remedies, including removing the foam themselves, buying second-hand machines online or simply going without the therapy.
PHILIPS RECALLS SLEEP APNEA MACHINES, VENTILATORS OVER POSSIBLE CANCER RISK
The devices are called continuous positive airway pressure, or CPAP, machines. They force air through a mask to keep passageways open during sleep.
Untreated sleep apnea can cause people to stop breathing hundreds of times per night, leading to dangerous drowsiness and increased heart attack risk. The problem is more common in men than women, with estimates ranging from 10% to 30% of adults affected.
Most patients are better off using a recalled device because the risks of untreated sleep apnea still outweigh the potential harms of the disintegrating foam, physicians say. But doctors have been hard pressed to help patients find new machines, which generally cost between $500 and $1,000, and were already in short supply due to supply chain problems.
"What happened is the company just said, ‘Talk to your doctor.’ But doctors can’t manufacture new machines out of the blue," said Dr. John Saito, a respiratory specialist near Los Angeles.
Risks from the foam include headache, asthma, allergic reactions and cancer-causing effects on internal organs, according to the Food and Drug Administration. The recalled devices include Dreamstation and SystemOne CPAP models and several other Philips machines, including Trilogy ventilators.
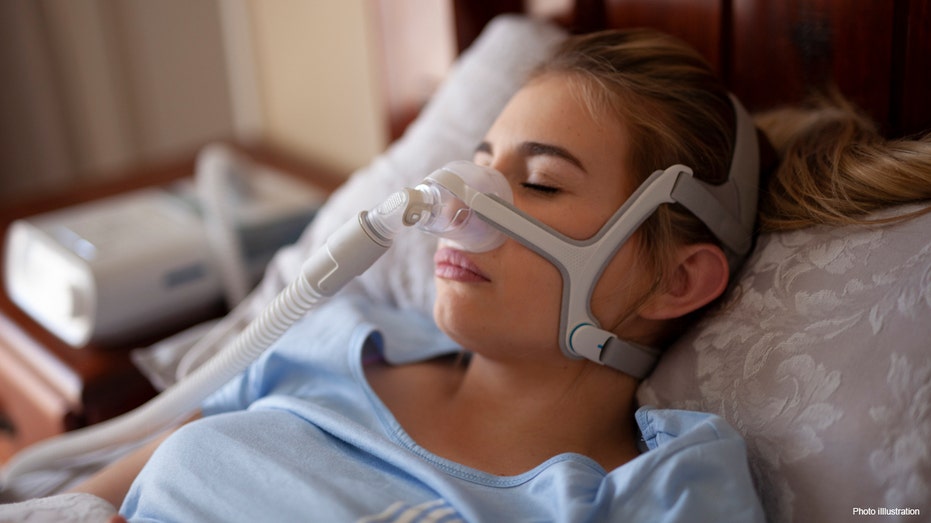
Cpap for Sleep Apnea on Woman in Bedroom (istock / iStock)
Last March, the FDA took the rare step of ordering Philips to expand its communication effort, including "clearer information about the health risks of its products." Regulators estimated then that only half of U.S. consumers affected had registered with the company.
The agency hadn’t issued such an order in decades.
In a statement, Philips said ongoing testing on the recalled devices is "encouraging" and shows low levels of particles and chemical byproducts emitted by its leading brand of machine. Philips said its initial communication about the dangers posed by the foam was "a worst-case scenario for the possible health risks." The deterioration appears to worsen with unauthorized cleaning methods, the company noted.
The FDA has received more than 70,000 reports of problems attributed to the devices, including pneumonia, infection, headache and cancer. Such reports aren't independently confirmed and can't prove a causal connection. They can be filed by manufacturers, patients, physicians or attorneys.
PHILIPS TO CUT 4K JOBS WORLDWIDE AMID FALLING SALES
Jeffrey Reed, of Marysville, Ohio, had been using his Philips machine for about a year when he began seeing black specks in the tubing and mask. His equipment supplier said the debris was caused by improper cleaning, so he continued using it.
Over the next seven years, Reed says he experienced persistent sinus infections, including two bouts of pneumonia, that didn’t resolve with antibiotics. After hearing about the recall, he suspected the foam particles might be playing a role.
"Once I got off their machine, all of that cleared right up," said Reed, 62, who obtained a competitor's device after several months. Like other users, Reed can’t definitively prove his problems were caused by Philips' device.
More than 340 personal injury lawsuits against Philips have been consolidated in a Pennsylvania federal court and thousands more are expected in coming months. Reed isn't part of the litigation.
Like the vast majority of U.S. CPAP users, Reed got his device through a medical equipment supplier contracted by his insurer. The company went out of business before the recall and he never heard from them about a replacement.

Jeffrey Reed, who experienced persistent sinus infections and two bouts of pneumonia while using a Philips CPAP machine, poses with the device at his home Thursday, Oct. 20, 2022, in Marysville, Ohio. The device is part of a massive global recall of ((AP Photo/Jay LaPrete) / AP Images)
Even in normal circumstances, those companies typically don’t track patients long term.
"After a couple years, you’re just forgotten in the system," said Ismael Cordero, a biomedical engineer and CPAP user. "I stopped hearing from my supplier about three years after I got my machine."
Cordero learned that his Philips machine had been recalled through his work at ECRI, a nonprofit that reviews medical device safety.
In May, the FDA put Philips on notice that it was considering a second order that would force the company to improve and accelerate its repair-and-replace program.
Medical device companies typically conduct recalls voluntarily, and former FDA officials say the agency has never actually used its authority to force additional steps.
"The FDA shares the frustrations expressed by patients who are awaiting a resolution for this recall," the agency said in a statement. Philips still hasn't provided "all information we requested to evaluate the risks from the chemicals released from the foam."
Philips disclosed earlier this year that it received a Department of Justice subpoena over the recall. The agency hasn’t publicly commented on the matter, per federal rules.
But an FDA inspection of Philips' Pennsylvania offices uncovered a spate of red flags last fall, including emails suggesting the company was warned of the problem six years before the recall. In an October 2015 email, one customer appeared to warn Philips that the polyester polyurethane foam could degrade, according to FDA.
Between 2016 and early 2021, FDA found 14 instances where Philips was made aware of the issue or was analyzing the problem internally. "No further design change, corrective action or field correction was conducted," the FDA inspectors repeatedly note.

Philips office building in Warsaw, Poland on July 29, 2021. (Photo by Beata Zawrzel/NurPhoto via Getty Images) (Beata Zawrzel/NurPhoto via Getty Images / Getty Images)
In a May 2018 email, foam supplier William T. Burnett wrote to Philips in an email: "We would not recommend use of polyester foam in such an environment. ... It will eventually decompose to a sticky powder," according to an affidavit filed as part of a lawsuit over the foam.
Since the recall, Philips has been using a new type of foam made from silicone to refurbish machines.
But FDA alerted consumers last November that the new material had failed one safety test. And regulators asked the company to perform more testing to clarify any health risks with both the new foam and the recalled material. Philips says independent testing has not identified any safety issues.
The company says it has replaced or repaired about 69% of recalled devices globally and aims to ship 90% of those requested by year’s end. On average, the company produces about 1 million sleep devices annually.
"We have scaled up by more than a factor of three, but inevitably it still takes time to remediate 5.5 million devices globally," the company said. About half are in the U.S.
Jeffrey Reed is among those still waiting.
Reed registered for a replacement device in June 2021 — within a week of the recall. This month, he received an email from Philips indicating that his device has been discontinued and isn't available for immediate replacement. Instead, the company offered him $50 to return the machine or an option of providing additional information to get a newer one.
CLICK HERE TO GET FOX BUSINESS ON THE GO
"For them to wait until October to tell me that my machine is too old, when they’ve known exactly what device I have since the day I registered — that’s frustrating," Reed said. "It’s disappointing that a provider of life-saving equipment treats people like this."